Sickle cell disease (SCD) is a terminal disease resulting from sickled blood cells caused by an abnormally folded beta-globin chain. The sickled cells cause occlusion and hyperviscosity leading to a myriad of complications. Unfortunately, some stigma associated with SCD persists in the healthcare field leading to undertreatment of pain or marginalization of patients. The American Society of Hematology (ASH) currently has draft recommendations that are open for public comment until May 13, 2019.
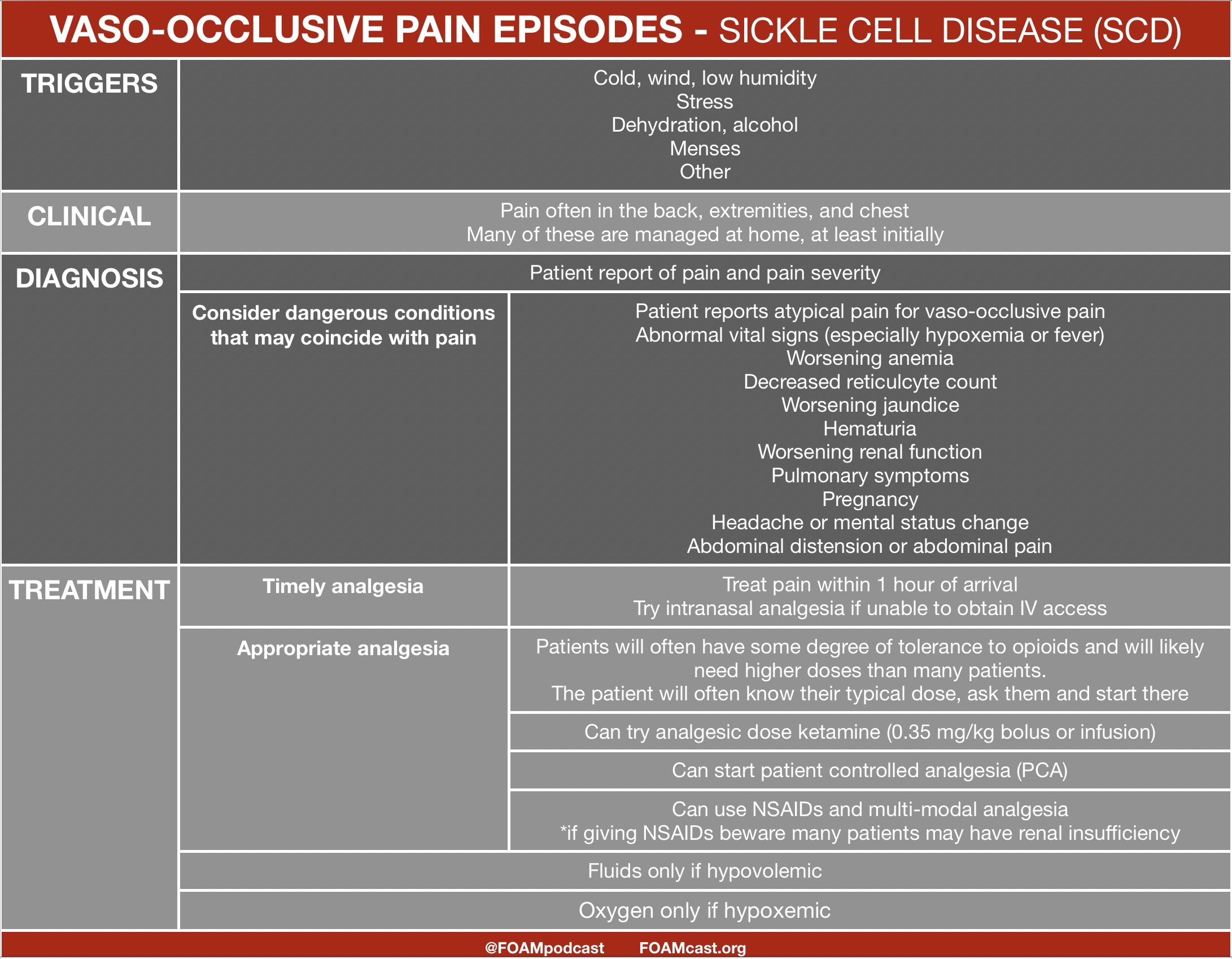

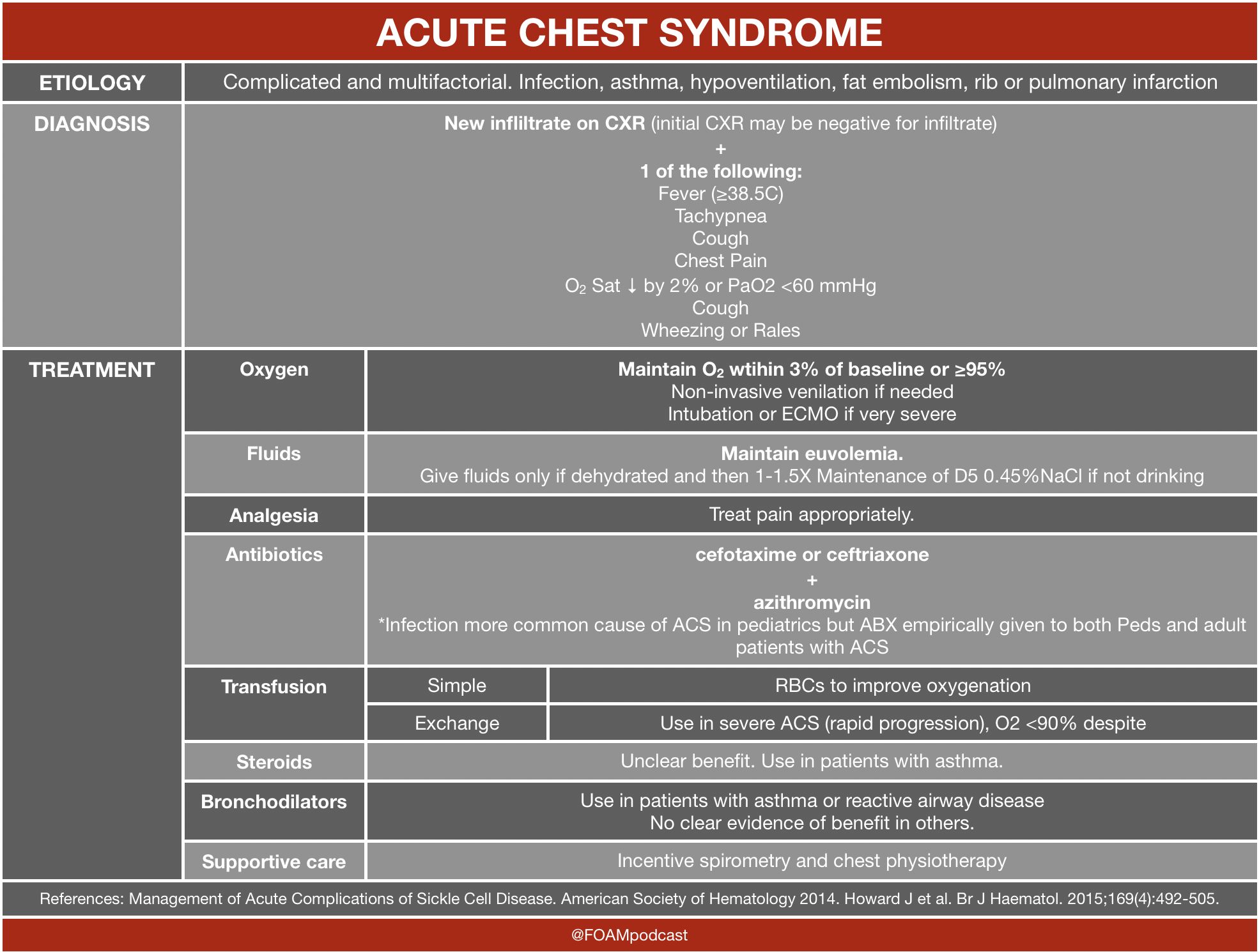

![]()
Which of the following is the most common cause of an aplastic crisis in a child with sickle cell disease?
A. Epstein-Barr virus
B. Parvovirus B19
C. Salmonella
D. Streptococcus pneumoniae
[accordion]
[toggle title=”Answer” state=”closed”] B. Predominantly affecting individuals of African ancestry, sickle cell anemia is a genetic disease that results in the formation of sickled red blood cells. Affected patients are homozygous for sickle hemoglobin (HbSS) which results in deoxygenated red blood cells developing a sickle or crescent shape. This leads to inflexible red blood cells, increased blood viscosity, and decreased blood flow within organs or extremities. A complication of sickle cell anemia is a transient aplastic crisis. This is caused by a short-lived stoppage of erythropoiesis, resulting in acute reductions in red cell precursors in the bone marrow, severely reduced reticulocytes in the peripheral blood, and an abrupt drop in hemoglobin level. Return to normal erythropoiesis usually occurs within two to 14 days. This transient aplastic crisis is typically caused by infection, with Parvovirus B19 being the most common etiologic agent in children. Individuals with a transient aplastic crisis are managed with transfusion. [/toggle]
[/accordion]
References:
- Glassberg J, Tanabe P, Richardson L, Debaun M. Among emergency physicians, use of the term “Sickler” is associated with negative attitudes toward people with sickle cell disease. Am J Hematol. 2013;88(6):532-3.
- Aisiku IP, Smith WR, Mcclish DK, et al. Comparisons of high versus low emergency department utilizers in sickle cell disease. Ann Emerg Med. 2009;53(5):587-93.
- Lovett PB, Sule HP, Lopez BL. Sickle Cell Disease in the Emergency Department. Hematol Oncol Clin North Am. 2017;31(6):1061-1079.
- Glassberg JA, Tanabe P, Chow A, et al. Emergency provider analgesic practices and attitudes toward patients with sickle cell disease. Ann Emerg Med. 2013;62(4):293-302.e10.